The emergence of infectious diseases has devastating socio-economic, health, and food security consequences. Our ultimate goal is to determine how evolution drives infectious disease emergence through the diversification of critical virulence factors. To accomplish this goal, we use a multidisciplinary approach that combines microbiology, experimental evolution, comparative genomics, and bioinformatics. By identifying and characterizing genomic changes that drive disease emergence, we will be able to more effectively forecast outbreaks and engineer more durable resistance in agriculture and medicine.
What determines the fate of host-pathogen interactions?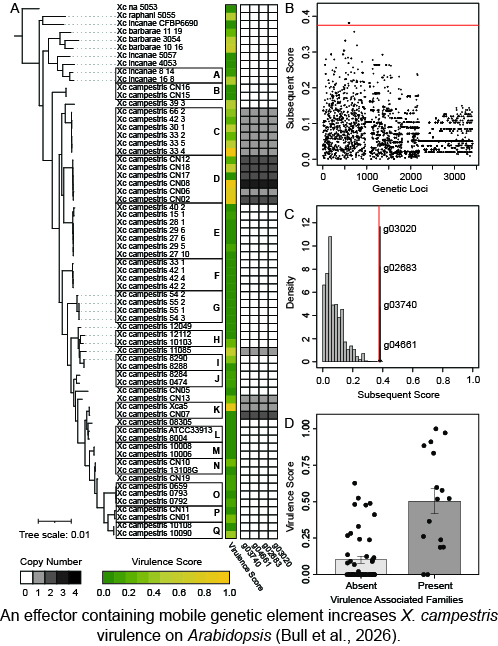
Most pathogens have a restricted host range and can only cause disease on a small subset of the hosts that they encounter. The factors that create these barriers have puzzled plant pathologists for decades. We are extending a pangenome approach that we developed in the model Pseudomonas syringae pathosystem to study the evolution of type III secreted effectors and other virulence factors in a collection of critical pathogens from the Xanthomonas genus. We will identify clusters of effector diversity that have resulted from the ongoing evolutionary arms race between Xanthomonas effectors and host immunity. We will then characterize host specificity determining effectors and ultimately develop a probabilistic framework for predicting host-pathogen associations through their effector repertoires.
How do novel infectious diseases emerge?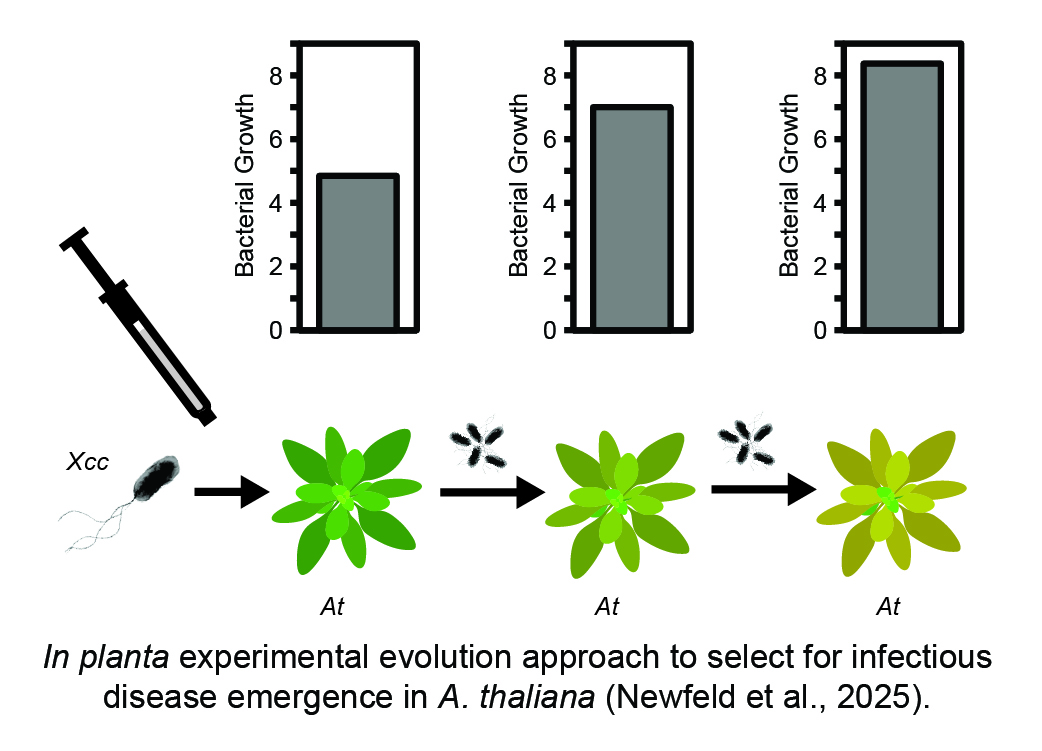
Experimental evolution provides an unbiased screen for adaptive mutations that drive infectious disease emergence. We are leveraging a novel in planta experimental evolution approach to select initially avirulent bacterial strains to the model host Arabidopsis. These lineages are monitored via time course sequencing and fitness assays to characterize adaptations at the genetic and phenotypic levels. Ultimately, we will explore how genetic background, chance, and selection contribute to the probability of a host shift and identify evolutionary trade-offs associated with these shifts.
How does the population genetic environment impact AMR?

The emergence of antimicrobial resistance (AMR) in clinical pathogens represents a growing public health crisis that threatens to diminish our ability to treat infectious diseases. We are performing hundreds of parallel, high-throughput evolve-and-resequence experiments to explore the impact of effective population size (Ne), antimicrobial concentration, and bacterial physiology on AMR evolution. Through this work, we have shown that evolution at higher Ne selects for stronger resistance phenotypes via a more predictable suite of mutations. As we expand this work to other population genetic factors, we aim to help guide the improved deployment of antimicrobial therapies that limit the evolution of broadly resistant bacteria.
Other Interests
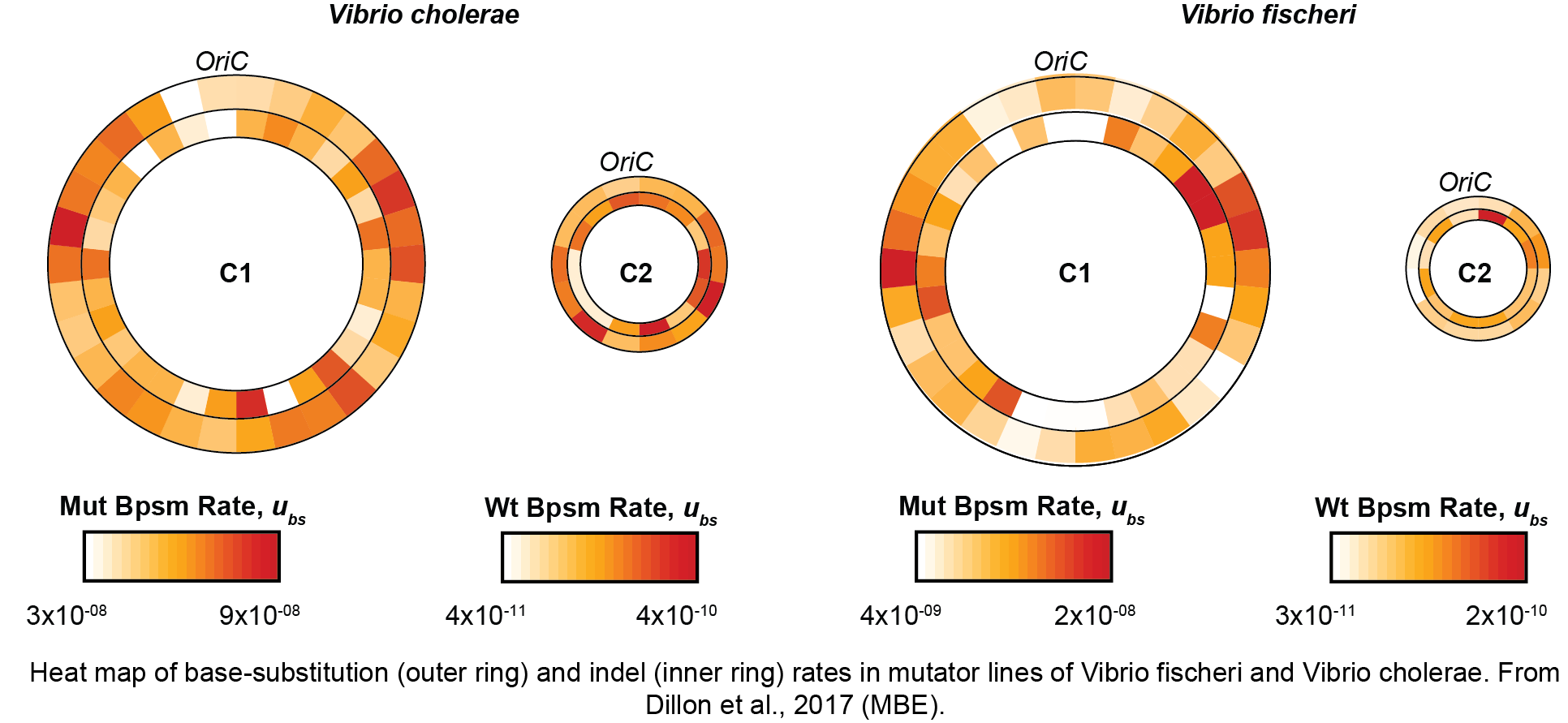
In the Dillon Lab, we are also broadly interested in basic questions about the rates and effects of spontaneous mutations. Despite having a fundamental importance for all of evolution, we’ve only recently begun to understand how mutation rates and fitness effects vary within and between genomes. Our work has shown that mutation rates vary in a wave-like pattern around larger primary bacterial chromosomes, but that this effect is diminished on smaller secondary chromosomes. These results have led us to hypothesize that patterns of periodic variation in mutation rate are related to replication timing. We’ve also demonstrated that most mutations have little or no effect on fitness, but those that do are mostly deleterious across multiple environments. We are currently pursuing several follow-up projects related to this work, including quantifying the mutation rates in repetitive transcription activator-like effectors and exploring the rates of chromosome fusion in bacteria with multiple chromsomes.